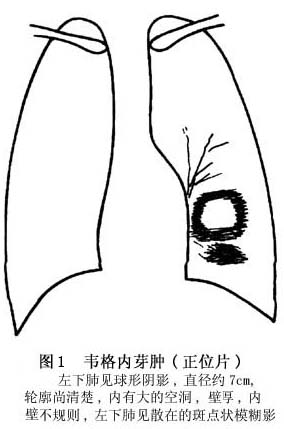
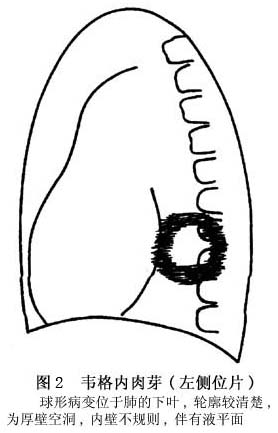
ЗЂВЁЛњжЦ
ЗЂВЁЛњжЦ
ЗЂВЁЛњжЦЃКФПЧАгаЯрЕБЖрЕФжЄОнжЇГжЮЄИёФЩШтбПжзВЁЪЧвЛИіздЩэУтвпадМВВЁЃЌANCAПЩФмВЮгыСЫбЊЙмЕФМЄЛюКЭЫ№ЩЫЁЃШчWGгыПЙPR3ЕФздЩэПЙЬхгаЧПЕФЬивьЙиЯЕЃЌПЙЬхаЇМлгыСйДВМВВЁЛюЖЏадЯрЙиЃЌВЂПЩдЄЪОИДЗЂЁЃМВВЁЖдУтвпвжжЦМСжЮСЦЗДгІСМКУЁЃЕЋвВгаВЛжЇГжЕФвРОнЁЃОЁЙмдкДѓВПЗжWGВЁШЫбЊЧхжаПЩМьВтЕНПЙPR3ЕФЬивьПЙЬхЃЌЕЋШдгаЩйВПЗжВЁШЫANCAвѕадЁЃЦфДЮЃЌдкЪмРлзщжЏжаЃЌМШУЛгаЗЂЯжздЩэПЙЬхЃЌвВУЛгаздЩэЗДгІадTЯИАћЃЌУЛгаЗЂЯжПЙPR3ЕФУтвпИДКЯЮяЁЃвђДЫЃЌЬсЪОМДЪЙANCAдкWGЕФжТВЁжагавЛЖЈзїгУЃЌвВВЛЪЧзюЛљБОЕФзїгУЁЃ
1.ANCAжТВЁЛњжЦ
(1)ANCAгыЖраЮКЫСЃЯИАћ(PMN)жЎМфЕФЗДгІЃКPMNдкБЛANCAМЄЛюЧАашЦфЫћбзЧАвђзгЕФЦєЖЏ(primed)ЃЌдкЯИАћБэУцБэДяКмЖрАћНЌПЙдЃЌАќРЈPR3КЭMPOЃЌЪЙЯИАћЛёЕУгыздЩэПЙЬхЯрЛЅзїгУЕФАаПЙдЁЃвбЦєЖЏЕФPMNдкгыANCAЯрЛЅзїгУКѓЃЌЪЧШчКЮБЛМЄЛюЕФШдгаељТлЁЃKettritzШЯЮЊЯИАћБэУцБэДяЕФPR3КЭMPOжЎМфЕФНЛСЊЪЧМЄЛюЕФЛљДЁЃЌвђЮЊANCAЕФF(abЁЏ)2ЦЌЖЮФмМЄЛювбЦєЖЏЕФPMNЃЌЖјFabЦЌЖЮдђВЛФмЁЃЦфЫћзїепЮДФмжЄЪЕЩЯЪіЗЂЯжЃЌЕЋЗЂЯжЭЈЙ§ПЙЬхгыFcЪмЬхЯрЛЅзїгУвВПЩМЄЛюPMNЃЌFcІУRЂђaКЭFcІУЂѓRbОљВЮгыСЫетИіЙ§ГЬЁЃСэЭтЃЌеыЖдІТ2ећКЯЫиЃЌЬиБ№ЪЧCD18ЕФЗтБеПЙЬхФмвжжЦANCAгеЕМЕФPMNМЄЛюЁЃ
МЄЛюЕФPMNПЩВњЩњЖОадбѕЛљЃЌЭбПХСЃЪЭЗХШмУИЬхУИЁЃСэЭтЃЌЫќУЧвВПЩЗжУкбзжЂНщжЪШчTNFІСЃЌIL-1ЃЌIL-8КЭLTB4ЁЃМЄЛюЕФPMN№ЄИНЗжзгБэДядіМгЃЌетЪЙPMNвзгкНсКЯВЂДЉЭИФкЦЄЯИАћВуЁЃWGВЁШЫЩіЛюМьБъБОЯдЪОЩіаЁЧђГіЯжМЄЛюЕФPMNЃЌЧвМЄЛюЕФPMNЪ§ФПгыЩіЙІФмЫ№КІЕФГЬЖШЯрЙиЁЃДЫЭтЃЌМЄЛюЕФPMNвВГіЯжгкбЊвКбЛЗжаЃЌМЄЛюЕФГЬЖШгыМВВЁЕФЛюЖЏадЯрЙиЁЃ
(2)ANCAгыЕЅКЫЯИАћжЎМфЕФЯрЛЅзїгУЃКANCAФмМЄЛюЕЅКЫЯИАћЃЌЪЙЦфЖОадбѕЛљЃЌIL-8КЭMIP-1ВњЩњдіМгЁЃМЄЛюЧАВЛашвЊЦєЖЏЃЌЕЋЦєЖЏФмЬсИпANCAНщЕМЕФЖОадбѕЛљЕФВњЩњЁЃ
(3)ANCAгыФкЦЄЯИАћжЎМфЕФЙиЯЕЃКФкЦЄЯИАћЪЧЗёБэДяANCAЕФАаПЙд(ЬиБ№ЪЧPR3)ШдгаељТлЁЃФкЦЄЯИАћдкбзЧАвђзгЕФДЬМЄЯТЃЌPR3БэДядіМгЃЌВЂДгАћжЪзЊЮЛЕНЯИАћФЄЩЯЃЌЪЙPR3ФмгыANCAЯрЛЅзїгУЁЃPR3-ANCAФмгеЕМФкЦЄЯИАћ№ЄИНЗжзгЕФЩЯЕїКЭIL-1ЁЂзщжЏвђзгЕФБэДяЁЃФкЦЄЯИАћгыPR3-ANCAЗѕг§ЪБЃЌФкЦЄЯИАћКЯГЩЧАСаЛЗЫиЁЂPAFЁЂIL-8діМгЃЌЕААзЩјТЉдіМгЃЌФкЦЄЯИАћЕђЭіЁЂЭбТфКЭШмНтЁЃ
(4)злЩЯЫљЪіЃЌВЮгыANCAЯрЙибЊЙмбзЕФЛњжЦШчЯТЃК
ЂйгЩгкОжВПИаШОЖјЪЭЗХЕФЯИАћвђзгв§Ц№ФкЦЄЯИАћ№ЄИНЗжзгЕФЩЯЕїЃЌВЂЦєЖЏжаадСЃЯИАћКЭ(Лђ)ЕЅКЫЯИАћЁЃ
ЂкбЛЗжавбЦєЖЏЕФжаадСЃЯИАћКЭ(Лђ)ЕЅКЫЯИАћдкЦфЯИАћБэУцБэДяANCAПЙдЁЃ
ЂлвбЦєЖЏЕФжаадСЃЯИАћКЭ(Лђ)ЕЅКЫЯИАћ№ЄИНгкФкЦЄЯИАћЃЌЫцКѓБЛANCAМЄЛюЁЃМЄЛюЕФжаадСЃЯИАћКЭ(Лђ)ЕЅКЫЯИАћЪЭЗХЖОадбѕЛљКЭШмУИЬхУИЃЌЫќУЧЕМжТФкЦЄЯИАћЫ№ЩЫЃЌзюжеЕНЛЕЫРадбзжЂЁЃ
ЂмANCAМЄЛюЕФжаадСЃЯИАћКЭ(Лђ)ЕЅКЫЯИАћЭбПХСЃЪЭЗХЕААзУИ3КЭЫшЙ§бѕЛЏЮяУИЃЌPR3КЭMPOЪЙФкЦЄЯИАћМЄЛюЁЂЫ№ЩЫЩѕжСЕђЭіЁЃЦфДЮЃЌвбНсКЯПЙЬхЕФPR3КЭMPOзїЮЊжжжВПЙдЃЌдкдЮЛаЮГЩУтвпИДКЯЮяЃЌШЛКѓдйЮќв§ЦфЫћжаадСЃЯИАћЁЃ
ЂнANCAМЄЛюЕФЕЅКЫЯИАћВњЩњMCP-1КЭIL-8ЃЌетаЉЧїЛЏЮяжЪЕФЪЭЗХПЩРЉДѓЕЅКЫЯИАћКЭжаадСЃЯИАћФММЏЕФГЬЖШЃЌПЩФмЕМжТШтбПжзаЮГЩЁЃ
2.ИаШО ИаШОвђЫивВПЩФмЕМжТWGЃК
(1)КмЖрWGВЁШЫЕФГѕЪМжЂзДгыИаШОадМВВЁЯрЫЦЃЌВЁШЫГЃГЃвђЮЊКєЮќЕРжЂзДОЭеяЁЃ
(2)дкWGВЁШЫжаНјааЕФжЇЦјЙмЗЮХнЙрЯДЯдЪОЃЌВЁШЫЭЈГЃБэЯжЮЊжаадСЃЯИАћЗЮХнбзЁЃ
(3)вбжЊгаМИжжИаШОгыФГаЉРраЭЕФбЊЙмбзЯрЙиЁЃдкШЫРрЃЌбЊЙмбзЕФЗЂЩњгывваЭИЮбзЁЂ
БћаЭИЮбзЁЂEpstein-BarrВЁЖОЁЂparvo-B19КЭHIVИаШОЯрЙиЁЃШЛЖјНігаВЛзу1%ЕФИаШОЛМепЗЂЩњбЊЙмбзЃЌЬсЪОЫожїЕФЬиеїОіЖЈСЫМВВЁЕФБэДяЁЃ
(4)SubraБЈИцСНР§ЛМбЧМБадаФФкФЄбзЕФВЁШЫЃЌC-ANCAОљбєадЁЃвЛЧ¨ШЫОПЙЩњЫижЮСЦНЕЕЭСЫC-ANCAЕФаЇМлЁЃСэвЛЧ¨ШЫОПЙЩњЫиКЭЭтПЦжЮСЦЪЙC-ANCAЯћЪЇЁЃЕЋФГаЉШЫШЯЮЊЃЌНЋГжајИаШОзїЮЊбЊЙмбзЕФДЬМЄМСЕФРэТлЪЧеОВЛзЁНХЕФЃК
ЂйжБЕННёЬьЃЌЦјЕРЛюМьБъБОЕФзщжЏВЁРэбЇбаОП(АќРЈЮЂЩњЮяЕФЬиЪтШОЩЋКЭЯИОњЁЂПЙЫсИЫОњЁЂецОњЁЂжЇдЬхКЭКєЮќЕРВЁЖОЕФХрбј)ЖМУЛгаФмЙЛжЄЪЕжТВЁЮЂЩњЮяЕФДцдкЁЃ
ЂкдквваЭИЮбзЁЂ
БћаЭИЮбзЯрЙибЊЙмбзЕФВЁШЫжаЃЌгІгУУтвпвжжЦжЮСЦКѓЃЌВЁЧщЫфгаЯджјИФЩЦЃЌЖјЫћУЧаЏДјЕФВЁЖОШДУїЯддіМгЁЃ
НќФъРДЃЌЗЂЯжЮЄИёФЩШтбПжзВЁгыСНжжЬиЪтЕФЮЂЩњЮяЯрЙиЃКЮЂаЁВЁЖО(parvovirus)B19КЭН№ЛЦЩЋЦЯЬбЧђОњЁЃStegemanЗЂЯжБЧЧЛН№ЛЦЩЋЦЯЬбЧђОњЕФГЄЦкДјОњгыМВВЁЕФИДЗЂЯрЙиЁЃН№ЛЦЩЋЦЯЬбЧђОњаЏДјепИДЗЂТЪЮЊЮоаЏДјепЕФ8БЖЁЃгІгУTMP/SMXКѓПЩНЕЕЭЛКНтЦкВЁШЫЩЯЦјЕРКЭБЧЧЛЕФИДЗЂЁЃЦЯЬбЧђОњВњЩњЕФГЌПЙд(SAg)ПЩФмЪЧWGЕФвЛИіживЊЕФДЅЗЂвђзгЃЌSAgМШПЩМЄЗЂздЩэЗДгІTЯИАћЃЌвВПЩМЄЛюздЩэЗДгІадBЯИАћЃЌВЮгыбЊЙмбзЕФВЁРэЩњРэЁЃFinkelдк1Р§WGВЁШЫжаЃЌЗЂЯжПЙ-B19ЃЌIgMЗДгІГжајНќ4ФъЃЌгІгУГВЪНPCRжЄЪЕЮЊВЁЖОбЊжЂЃЌОВТізЂЩфУтвпЧђЕААзЃЌВЁШЫжЂзДЯджјИФЩЦЁЃ
3.ШтбПжзаЮГЩЛњжЦ дкЦфЫћМВВЁжаЃЌШтбПжзЭЈГЃЪЧгЩжТУєЕФCD4 TЯИАћ(ПЩВњЩњThlЯИАћвђзг)НщЕМЕФЁЃдкWGжавВГіЯжЯрЫЦЕФбзжЂЃЌгавЛжжМйЫЕШЯЮЊЃЌзщжЏЫ№ЩЫКЭбЊЙмбзЪЧЗёгЩЛћБф(ВЛе§ГЃ)ЕФThlУтвпЗДгІНщЕМЕФЁЃгаМИЯюбаОПжЇГжетИіМйЫЕЃКдкWGКЭЯрЙиЕФбЊЙмбзжаЃЌЯИАћвђзгЕФВњЩњгаЖЈадЁЂЖЈСПЕФвьГЃЁЃдкWGВЁШЫжаЃЌбЊЧхIL-1ЁЂIL-2ЁЂIL-6КЭTNF-ІСЫЎЦНЩ§ИпЃЌбЛЗЕЅКЫЯИАћTNF-ІСЕФВњЩњдіМгЁЃгІгУЗДзЊТМОлКЯУИСЊЗДгІ(RT-PCR)ЁЂдЮЛдгНЛКЭУтвпзщЛЏММЪѕЗЂЯжВЁШЫЩіаЁЧђIL-1КЭTNF-ІСЕФВњЩњдіМгЁЃзюНќLudvikssonЭЈЙ§баОПЛюЖЏЦкWGВЁШЫЕФЭтжмбЊСмАЭЯИАћЃЌЗЂЯжгые§ГЃШЫЯрБШЃЌЛМепCD4 TЯИАћВњЩњЕФIFN-ІУЫЎЦНИп10ЁЋ20БЖЃЌTNF-ІСЕФВњЩњвВгаЯджјдіМгЁЃЯрЗДЃЌTh2ЯИАћвђзг(IL-4ЁЂIL-5ЛђIL-10)ЕФЫЎЦНЮоЯджјВювьЁЃ LudvikssonЛЙЙлВьЕНЮоТлЪЧЛюЖЏЦкЃЌЛЙЪЧЛКНтЦкЃЌВЁШЫЕЅКЫЯИАћIL-12ЕФВњЩњОљдіМгЃЌЖјIL-12ЮЊTЯИАћЯђTh1ЯИАћ(ПЩВњЩњIFN-ІУ)ЕФЛљБОгеЕММСЁЃ
злЩЯЃЌЕБWGВЁШЫБЉТЖгкЛЗОГДЬМЄ(ШчИаШО)КЭ(Лђ)здЬхПЙдгеЕМЕФЙ§ЖШОоЪЩЯИАћIL-12ЗДгІЃЌв§Ц№Th1ЯИАћвђзг(TNF-ІСЁЂIFN-ІУ)ВњЩњдіМгЃЌTNF-ІСЁЂIFN-ІУЃЌПЩЦєЖЏВЂЮЌГжШтбПжзадбЊЙмВЁБфЁЃДЫЙ§ГЬПЩБЛANCAгАЯьЃЌANCAПЩДйНјжаадСЃЯИАћЁЂФкЦЄЯИАћКЭЕЅКЫЯИАћЕФМЄЛюЁЃ
СйДВБэЯж
СйДВБэЯжЃКвРСйДВБэЯжПЩЗжЮЊ3аЭЃК
1.ОЕфаЭ БэЯжЮЊБЧёМбзЁЂжаЖњбзЃЌБЧбзАщБЧ№ЄФЄРЃбёЃЌПШЫдЃЌ
ПЉбЊКЭШЋЩэжЂзДЁЃ
2.БЉЗЂаЭ БэЯжЮЊбИЫйЕФЩіЁЂКєЮќЙІФмЫЅНпЁЃ
ЖрЗЂгкЧрзГФъФаадЁЃзюГЃЧжЗИЕФВПЮЛЪЧИББЧёМЁЂБЧбЪЧЛЁЂЦјЙм№ЄФЄМАЗЮМфжЪЃЌврПЩЧжМАЦЄЗєЁЂблЁЂаФЕШЖрЯЕЭГЖрЦїЙйЁЃвдЩЯЁЂЯТКєЮќЕРЪмРлзюЖрЃЌВЂГЃгыБЧбЪВПВЁБфЭЌЗЂЃЌШчЗЂШШЁЂЧхЬщЁЂПШЫдЁЂ
ПЉбЊЁЂЦјЖЬЁЂаиУЦЕШжЂзДМАЗЮФкНўШѓадвѕгАЕШЃЌБЧбЪВПжЂзДШчИББЧёМЬлЭДЁЂБЧЧЛ№ЄФЄРЃбёЁЂБЧГібЊЃЌжСЭэЦкБЧЙЧНўШѓаЮГЩААБЧЕШЁЃ
дМ50%ЛМепГіЯжЦЄЗєбЊЙмбзБэЯжЃЌЕфаЭЦЄЫ№ЮЊПЩДЅжЊЕФзЯёАЃЌЖдГЦЗжВМгкУцВПКЭЫФжЋЃЌЬиБ№ЪЧжтЁЂЯЅКЭЭЮВПЃЌГЃГіЯжЛЕЫРадЧ№еюЁЂЫЎ№хЁЂбЊ№хЕШЃЌХМПЩЗЂЩњЗчЭХЁЂКьАпЁЃЯТжЋдЖЖЫНЯЮЊУїЯдЃЌвВПЩЗЂЩњгкЦфЫћЙЧїРЭЛГіВПЮЛЁЃЫцВЁЧщНјеЙГіЯжЦЄЯТНсНкЃЌГЪШтКьЩЋЛђзЯЩЋЃЌжЪгВЃЌгаЧсЖШДЅЭДЃЌбЯжиепПЩгажабыЦЦРЃЁЂЛЕЫРЁЃЩѕЛђГіЯжИќбЯжиЕФЛЕОваЭЫ№ЩЫЁЃМБадБЉЗЂаЭЛМепПЩгаРзХЕеїЃЌжИЖЫОжВПШБбЊЛђЛЕЫРЃЌЩрЭјзДЧрАпЁЃ
ЩідрВЁБфГіЯждкВЁГЬжаЭэЦкЃЌШчЮДФмНјаагааЇжЮСЦПЩв§жТЩіЙІФмЫЅНпЃЌЪЧживЊЫРвђжЎвЛЁЃблВПВЁБфПЩгаНсФЄбзЁЂ
РсЯйбзЁЂЙЎФЄбзЁЂКчФЄбзЁЂ
ЪгЭјФЄбЊЙмбзЁЂблЧђЭЛГіЁЂЪгСІеЯАЕШЁЃ
ДЫЭтЃЌдМ90%ВЁШЫгагЮвЦадЯЅЁЂѕзЕШДѓЙиНкЬлЭДЛђЙиНкбзЃЌЕЋвЛАуВЛв§жТЙиНкЛћаЮЁЃдМ60%ВЁШЫгажаЖњбзЁЃдМ20%гаЩёОЯЕЭГЪмРлЃЌвдЕЅЗЂЛђЖрЗЂЩёОбзЖрМћЁЃдМ12%ВЁШЫгааФдрЪмРлЁЂГіЯжЙкзДЖЏТібзЕШЁЃГЄЦкБЧВПаЏДјН№ЛЦЩЋЦЯЬбЧђОњПЩдіМгМВВЁИДЗЂЕФЮЃЯеЁЃ
МјБ№еяЖЯ
МјБ№еяЖЯЃКБиаызЂвтгыЦфЫћОпгаШтбПжзадбзжЂЁЂбЊЙмбзМВВЁЯрМјБ№ЁЃ
1.ЗЮГібЊ-ЩібззлКЯеї ВЁРэбЇМьВщЪОЬхФкДцдкПЙЩіаЁЧђЛљЕзФЄПЙЬхМАгЋЙтПЙЬхМьВщгаЯпзДХХСаЕФIgGЃЌетгыЮЄИёФЩШтбПжзВЛЭЌЁЃ
2.СмАЭСібљШтбПжз Г§ЮоЩЯКєЮќЕРЪмРлЭтЃЌЩагаЩіДЉДЬЛюМьЪОЩіаЁЧђСмАЭбљНўШѓЃЌетаЉЖМПЩгыБОВЁМјБ№ЁЃ
3.ЬиЗЂаджаЯпШтбПжз ЪЧвЛжжУцВПКЭЩЯКєЮќЕРОжВПЦЦЛЕМВВЁЃЌЭЈЙ§ЩідрДЉДЬЛюМьПЩгыБОВЁМјБ№ЁЃ
4.ЦфЫћЮЄИёФЩШтбПжз Ащга
ЪШЫсСЃЯИАћдіЖржЂЪБЃЌЛЙгІзЂвтгыв§Ц№ЪШЫсСЃЯИАћдіЖрЕФаэЖрдЗЂВЁЯрМјБ№ЃЌгШЦфгІгыЙ§УєадШтбПжзЯрМјБ№ЃЌКѓепвдЗЂШШЁЂЗЂзїадЯјДЁЂЪШЫсСЃЯИАћдіЖрЮЊЬиеїЃЌЦфзщжЏВЁРэИФБфжївЊЮЊЪШЫсСЃЯИАћНўШѓЃЌВЁдюжмЮЇРрЩЯЦЄЯИАћГЪЗХЩфзДХХСаЃЌЪмРлЕФбЊЙмЮЊжаЁЂаЁЖЏТіЃЌЮЂЖЏТіЃЌОВТігыУЋЯИбЊЙмЃЌЮоИББЧёМбзЕФСйДВБэЯжКЭXЯпЬиеїЁЃСэЭтЃЌЮЄИёФЩШтбПжзЕФЗЮВПжЂзДМАXЯпЬиеївВгыЙ§УєадШтбПжзгаЯджјВЛЭЌЁЃ
жЮСЦ
жЮСЦЃКЩаЮоЬиЪтСЦЗЈЁЃжївЊЪЧдчЦкЗЂЯжКЭжЮСЦЁЃ
1.ФкгУжЮСЦ
(1)ЦЄжЪРрЙЬДММЄЫиЃКЮЊжЮСЦБОВЁЕФЪзбЁвЉЮяЃЌГЃгУСПЮЊЦУФсЫЩ1.0mg/(kgЁЄd)ЃЌЗжДЮПкЗўЁЃД§ВЁЧщЮШЖЈКѓж№НЅМѕСПжСЭЃвЉЁЃЮЊСЫдіЧПСЦаЇКЭМѕЩйЦфИБзїгУЃЌГЃгыЛЗСзѕЃАЗСЊКЯгУвЉЁЃ
(2)УтвпвжжЦМСЃКЪзбЁЛЗСзѕЃАЗЃЌГЃгыЦУФсЫЩСЊКЯгІгУЃЌЪЧЖдЛюЖЏадWegenerШтбПжзЕФзюКУЕФвЉЮяжЮСЦЗНАИЁЃПЩЪЙ90%ЛМепжЂзДЕУЕНЛКНтЁЃГЃгУСПЮЊ1ЁЋ2mg/(kgЁЄd)ЃЌЗж2ЁЋ3ДЮПкЗўЃЌШчЮЊБЉЗЂадЃЌПЩдіжС4mg/(kgЁЄd)ЃЌЛђОВТігУвЉЃЌвЛАугУжСМВВЁЛюЖЏЯћЭЫКѓМЬајЮЌГж6ИідТЕН1ФъЁЃгУвЉЪБгІзЂвтМрВтбЊЯѓЃЌЪЙАзЯИАћМЦЪ§ЮЌГждк3ЁС109/LвдЩЯЁЃвВПЩбЁгУСђпђрбпЪЁЃаТНќгаБЈЕРУПжмЕЭМССПМзАБЕћпЪ(15ЁЋ25mg)ПкЗўЛђМЁФкзЂЩфжЮСЦЃЌШЁЕУНЯКУСЦаЇЁЃ
(3)ПЙЩњЫиЃКгаМЬЗЂИаШОепгІбЁдёЪЪЕБПЙЩњЫиЁЃ
2.ОжВПжЮСЦ ПЩОнЦЄЫ№аджЪКЭРраЭбЁгУЪЪЕБЭтгУвЉЮяЁЃ
3.ЦфЫћжЮСЦ ОжВПЫ№КІПЩгУЗХЩфжЮСЦЁЂыВМЄЙтжЮСЦЕШЁЃБивЊЪБПЩааЩівЦжВЪѕЁЃ
дЄЗР
дЄЗРЃК
1.вЛМЖдЄЗР
(1)МгЧПгЊбјЃЌдіЧПЬхжЪЁЃ
(2)дЄЗРКЭПижЦИаШОЃЌЬсИпздЩэУтвпЙІФмЁЃ
(3)БмУтЗчКЎЪЊЃЌБмУтЙ§РлЃЌМЩбЬОЦЃЌМЩГдаСРБЪГЮяЁЃ
(4)ЪвЭтЛюЖЏЪББЃЛЄблгУблежЗРЛЄМАБЧВПЕФБЃЛЄЁЃ
2.ЖўМЖдЄЗР дчЦкеяЖЯЃЌСЫНтблЁЂБЧИаШОЧщПіЃЌзіКУСйДВЙлВьЃЌдчЦкЗЂЯжИїИіЯЕЭГЕФЫ№КІЃЌдчЦкжЮСЦЃЌжївЊПижЦблЁЂБЧЕФИаШОЁЃ
3.Ш§МЖдЄЗР зЂвтЗЮЁЂЩіЁЂаФМАЦЄЗєВЁБфЃЌВЂзЂвтМЬЗЂадН№ЛЦЩЋ
ЦЯЬбЧђОњИаШОЕФЗЂЩњЁЃДЫЭтЃЌЩёОЯЕЭГЁЂЯћЛЏЯЕЭГврПЩФмБЛРлМАЃЌгІгУжавЉПЩгаЕїНкУтвпЃЌЧхШШНтЖОЃЌЛюбЊЛЏ№іЕФЙІаЇЁЃ